Intro
A group at the Pacific Northwest Research Institute has published a new, annotated Mytilus trossulus genome, so I thought we’d go back and use that for our DGE analysis.
Steven aligned our tag-seq files to the new genome using HiSat and got slightly higher alignment rates compared with when we just used Kallisto to align to the iso-seq transcriptome.
We (Steven, Emily, and I) also thought it would make more sense to look at gene expression in treatments where there was actually a difference in byssus strength (the ocean warming and hypoxia treatments), and just to look at the foot tissue.
So, after Steven helped me put together the gene count matrix from our gtf files, I ran DESeq on foot tissue samples comparing the ocean warming and hypoxia treatments to our treatment control (day 3 in the lab).
Results
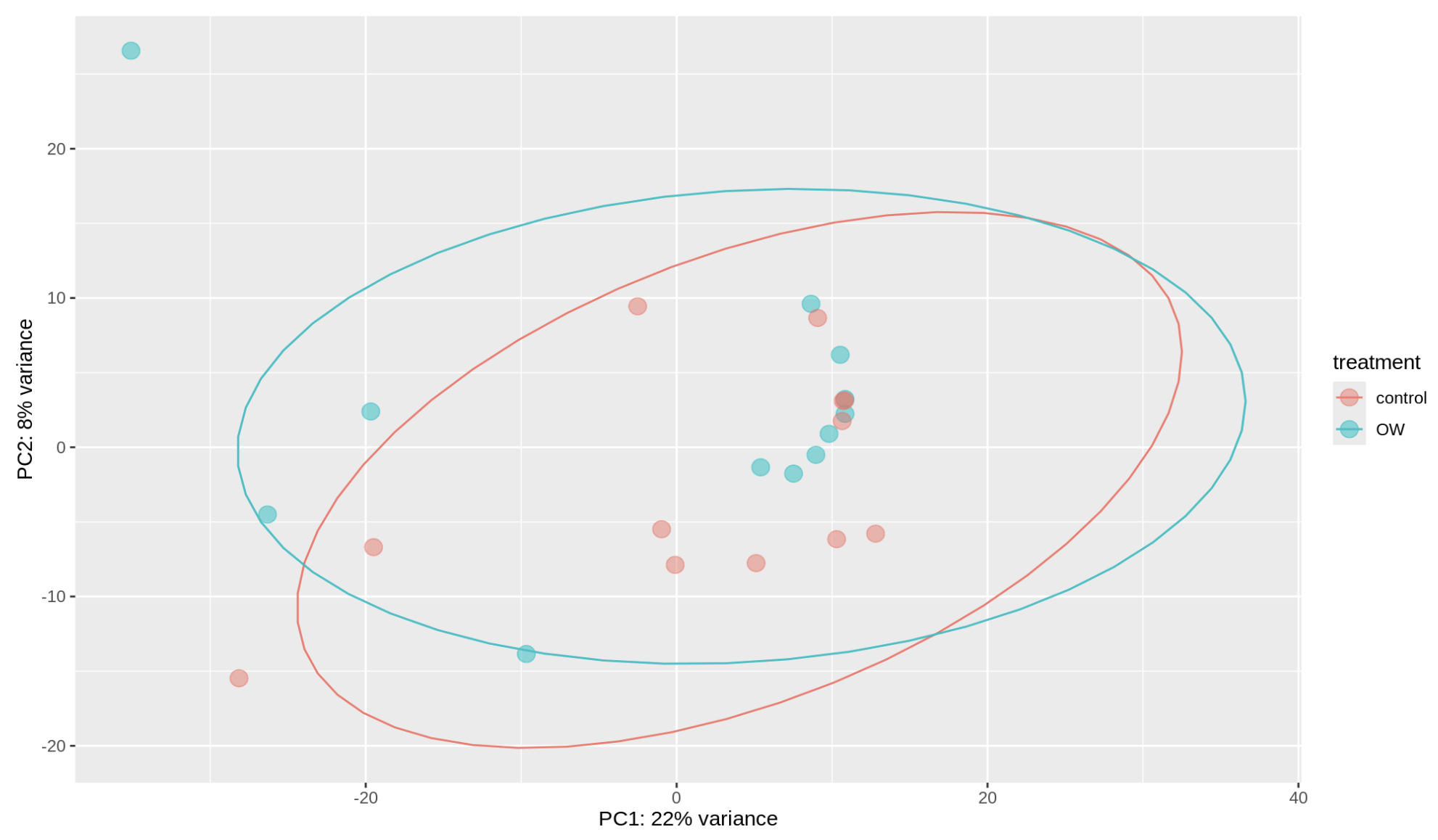
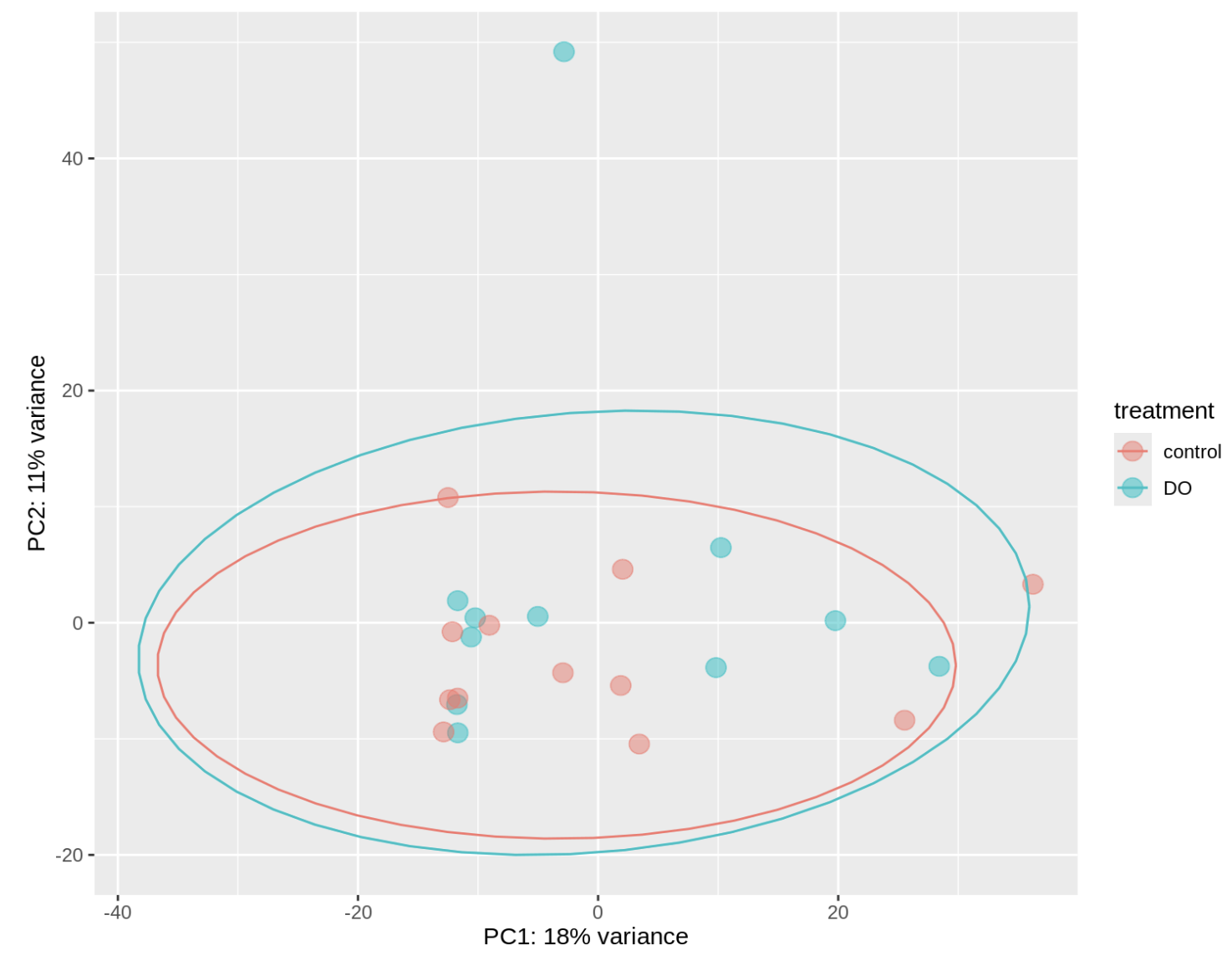
So we’re getting a different result here than when we aligned to the iso-seq transcriptome. The treatments are no longer visibly different from the control and much more variance is explained than before. There are some outliers which I’m not sure what to do with (are they worth removing?).
Next steps
Let me know if I should remove those outliers. This will determine whether I can move on to next steps.
Additionally, I got to the point where I filtered the data and put the data through shrinkage estimators, and I wanted to see how the genes mapped to GO terms but I’m not sure the old file Steven made that has the transcript ID and the GO terms works anymore, now that I have gene locations instead of unidentified transcripts.